
Author: Jessica McAnulty
Edited by: Alison Ludzki, Lihan Xie, and Sarah Kearns
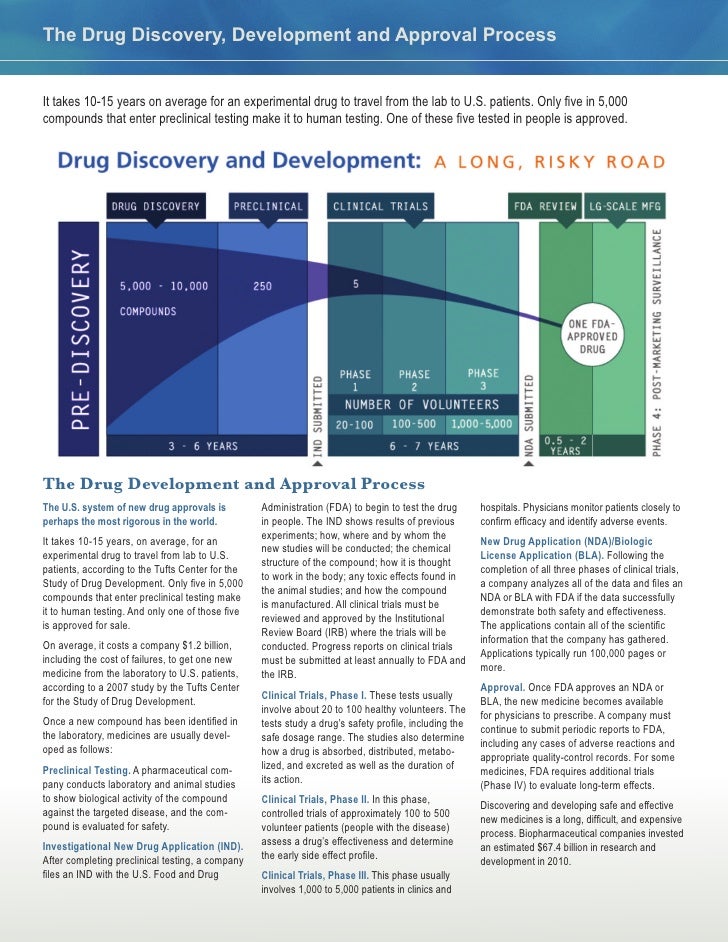
Take a look inside your medicine cabinet. Advil, Benadryl, Sudafed – ready at the snap of a finger if you fall ill. Developing that medicine, however, probably takes much longer than one would expect. On average, getting a potential drug candidate from the laboratory to the pharmacy takes about 14 years, costs more than one billion dollars, and has a low success rate. A successful drug will pass through all five stages: drug discovery, pre-clinical research, clinical trials, FDA approval, and post-market monitoring. Success statistics are gathered several stages into drug development; a new report states 13.8% of drug candidates that enter Phase I of clinical trials, the first test in humans, will earn FDA approval. Upon approval, companies have an exclusivity period ranging from 6 months to 7 years to earn back the large expense of drug development before generic medicines are released by competitors. For this reason, there is a necessary economic drive associated with pharmaceutical companies so that they can continue production of life-saving medication.
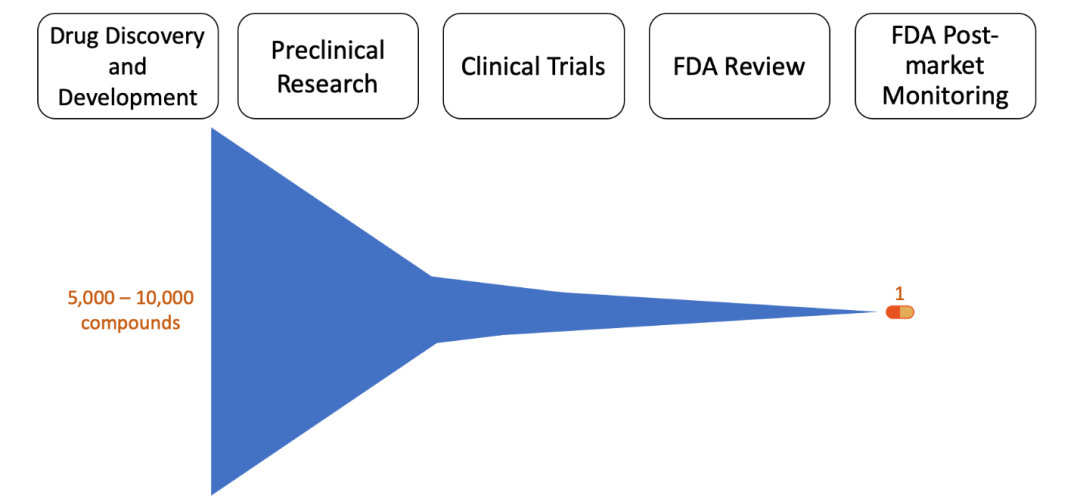 Figure 1, Stages of Drug Development: The blue figure represents the number of compounds that progress from the first to last stage of drug development over time.
Figure 1, Stages of Drug Development: The blue figure represents the number of compounds that progress from the first to last stage of drug development over time.
- Drug discovery:
This first stage, drug discovery, typically takes the most time and effort. A successful drug with few side effects needs to interact with a specific target, such as a protein or gene. However, it is rare for a disease to have a known target in the first place. First, scientists need an understanding of what drives the disease. Without this information, it is very difficult to predict a disease target and helps explain why scientists are still working on cures for diseases. Once molecular drivers are identified (outside of the drug discovery timeline), medicinal chemists and biologists work through a series of educated guesses to hopefully identify a target. If a target is known, scientists can screen through thousands of compounds (i.e. a drug screen) to find one that desirably affects the target. Scientists strategically select compounds to be used in the drug screen based off their structure-activity relationship (SAR), in which computers predict the biological effect based off the molecular structure of the compound by comparing it to similar compounds with known effects. It is important to note that, albeit more challenging, scientists can still perform a drug screen if the disease target is unknown. In this case, the drug screen is in search of compounds that prevent certain characteristics, or phenotypes, of the disease. If the drug screen successfully identifies compounds, scientists will conduct experiments with the compounds in cells grown on a plate (also known as in vitro experiments) to ensure the compound is effective in the cellular environment. Unfortunately, drug discovery can become cyclic if the initial compound has a poor potency (i.e. amount of drug to produce a positive effect) or is nonselective (i.e. any cell in the body is affected, rather than a specific target). A poorly potent, nonselective compound may have to be redesigned, and therefore repeat the drug screen process. Identifying a promising drug could take several attempts over 4-5 years. This stage also has the lowest success rate, with only ~250 out of 5,000-10,000 compounds moving to the next round of testing.
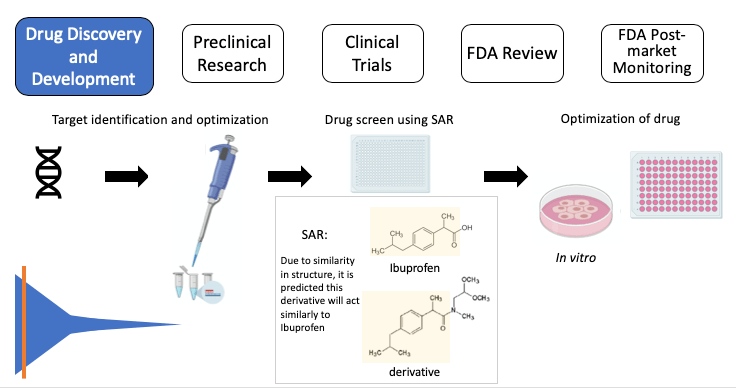 Figure 2, Drug Discovery: Target > target validation > drug screen using SAR: Ibuprofen (top), the active ingredient in Advil, and a derivative (bottom) share the chemical structure highlighted in yellow. SAR suggests the derivative will act similarly to ibuprofen > identify “hits” (successful compounds) > test in cells (in vitro) to further test the compound. SAR: structure-activity relationship. Note: some images in this graphic were created using BioRender.
Figure 2, Drug Discovery: Target > target validation > drug screen using SAR: Ibuprofen (top), the active ingredient in Advil, and a derivative (bottom) share the chemical structure highlighted in yellow. SAR suggests the derivative will act similarly to ibuprofen > identify “hits” (successful compounds) > test in cells (in vitro) to further test the compound. SAR: structure-activity relationship. Note: some images in this graphic were created using BioRender.
2. Pre-clinical studies:
When a compound shows promise in vitro, a proposal is submitted by the researcher to conduct research within a living organism, known as in vivo. The proposal is received by a committee at the research institution that strictly regulates animal research to ensure necessity and ethicality of experiments. Because of the physiological similarities in humans and animals, animal models are very important in aiding scientists in translating their findings to humans. These preclinical data help scientists understand dosage of the compound, side effects, differences between genders, and importantly, the effectiveness of the compound in treating the particular disease. The potency from the drug screen in the drug development phase will give researchers a ballpark on what the dosage should be, but that screen was performed in vitro. The dosage study is necessary in animals to determine if the drug has toxicities towards other cell types or organs, which would not be discovered in a test performed in cells. Pre-clinical studies take as long as a year to gather data and usually only about 5 of the 250 compounds that entered pre-clinical studies are successful. Some reasons a compound may fail include high required dosage, poor side effects, or inability to reach the target, such as passing through the blood-brain barrier (BBB*). Therefore, it is an incredible achievement for a compound to have success in an animal model and receive approval to be tested in humans through clinical trials.
(*The BBB is a membrane between the brain and its surrounding blood vessels. Although it is excellent at protecting the brain from disease-causing pathogens and toxins in the blood, it also prevents small molecules, like drug candidates, from reaching the brain.)
 Figure 3, Pre-clinical studies: Animal models aid in determining dosage and observing side effects, gender differences, and effectiveness compared to standard of care.
Figure 3, Pre-clinical studies: Animal models aid in determining dosage and observing side effects, gender differences, and effectiveness compared to standard of care.
3. Clinical trials:
Research in humans, just like in animals, requires extensive paperwork and approval. There are three phases in clinical trials, and as the compound moves to each phase, more people and checkpoints are added. Phase I enrolls 10-50 patients to ensure proper dosage in humans. Since this is the first test in humans, great attention is paid to observe any side effects; there is little concern of how the drug affects the disease as the primary focus is if the drug is tolerable. If the drug has too many side effects, it is not worth testing in sick patients. Estimates from 2018 suggest if a drug candidate passes Phase I, it has a 14% chance of obtaining approval. Phase II enrolls hundreds of patients with the specific disease they are aiming to treat. In addition to the potential drug, a placebo or “sugar pill”is implemented as a control. This helps scientists and doctors understand if their drug candidate is truly causing beneficial changes and it is not just a “placebo effect”. Last is Phase III, totaling about a thousand patients enrolled and may take several years to complete. Scientists are still looking out for side effects, significant differences in patient health compared to the placebo, and the new task of measuring if this new drug candidate is better than the standard of care (i.e. what is currently used to treat those patients). It is important to note that patients in clinical trials will always receive the placebo/drug candidate in addition to the standard of care. For example, the standard of care for many cancers is a type of chemotherapy. If there is a drug candidate for a certain cancer, it be given in combination with chemotherapy. However if no standard treatment exists, patients will only receive the experimental drug. Keep in mind it is possible for drug candidates to be rejected at any point in these clinical trials. For a drug to make it all the way to Phase III of clinical trials is extraordinary, and from here it has a 50% chance of getting approved. It is important to note that if a drug candidate proves early on that it was very effective for a life-threatening disease, the FDA will declare an accelerated approval and provide the drug to all enrolled patients.
 Figure 4, Clinical trials: As the drug candidate proceeds to the next phase, more patients are enrolled, and more characteristics are studied.
Figure 4, Clinical trials: As the drug candidate proceeds to the next phase, more patients are enrolled, and more characteristics are studied.
4. FDA Review:
Usually, only 14% of drugs that are tested in humans are submitted to the FDA for review. An application is submitted to the FDA, including all data that were generated over the decade. Review of a standard submission takes 10 months, but a candidate that received priority review will be reviewed in 6 months. During this time, the FDA reviews all data, including the drug label, and inspects the facility in which the drug will be made. During this inspection, officers inspect and observe records of the production process and collect samples to protect human safety. Upon approval, the FDA grants an exclusivity period, or exclusive marketing rights, ranging from 6 months to 7 years, depending on the category into which the new drug falls. Exclusivity can run concurrently with a patent, a property right issued by the United States Patent and Trademark Office that lasts 20 years from the date of application. Unlike exclusivity, a patent can be granted at any point during drug development.
 Figure 5, FDA Review: After successful completion of Phase III clinical trial, an application is submitted to the FDA with all collected data and a drug label. The FDA will inspect the drug manufacturing facility to ensure safety.
Figure 5, FDA Review: After successful completion of Phase III clinical trial, an application is submitted to the FDA with all collected data and a drug label. The FDA will inspect the drug manufacturing facility to ensure safety.
5. Post-market review:
Although the phases of clinical trials are long enough to monitor any short-term side effects, it is unknown how the drug will affect the body long-term. Phase IV of clinical trials begins after the drug received approval in order to monitor long term effects, observe if new side effects appear over time, and determine if the drug increases patients’ lifespans. This phase is especially important for drugs that received accelerated approval and therefore had shorter clinical trials.
 Figure 6, Post-market review: Continued Phase IV clinical trial to observe if there are any long-term side effects of the newly approved drug.
Figure 6, Post-market review: Continued Phase IV clinical trial to observe if there are any long-term side effects of the newly approved drug.
The cost to society:
A drug succeeding in FDA approval requires good predictions, planning, the right target identification, and resources. The costs, risks, and timeline of drug development are not commonly spoken of, so understandably, a patient who must pay a large sum for a necessary medicine may be upset. However, these costs must support the development of that particular drug, as well as the lost capital from previously failed attempts despite best efforts. Furthermore, once a patent and/or period of exclusivity expires, other companies can create and market a generic version of the FDA approved drug. Generic drugs are cheaper because drug discovery and clinical trials have already been conducted; a company producing a generic drug simply pays for the manufacturing of the pill once the generic is FDA approved. As a result of the lower cost to consumers, nearly 80% of prescribed drugs in the US are generics. For this reason, the original creator of the drug hopes to sell enough to match the cost of drug production before the competition begins.
Therefore, there is a necessary economic drive associated with big pharmaceutical companies; without money and resources to begin drug discovery, we wouldn’t have any medicines in the first place. Additionally, because of the major costs associated with drug development, it is difficult for pharmaceutical companies to investigate treatment for rare diseases. If a pharmaceutical company goes bankrupt producing a drug that helps a couple hundred people, then millions are left without medicines.
Arguably, there is an ethical decision that must be made. This predicament was realized and in 1983, the Orphan Drug Act was passed by Congress to provide financial incentives for pharmaceutical companies to pursue drug development for rare diseases. This act led to the creation of 250 FDA approved orphan drugs. Furthermore, there are examples of many individuals with a common disease, but do not have income or insurance to pay for medicine. Six pharmaceutical companies, including Eisai, GlaxoSmithKline, Merck & Co., Merck KGaA, Johnson & Johnson, and Pfizer, donated more than $22.3 billion of medicine to developing countries to help fight this problem.
Although medicine prices for the consumer are high, there is a need for large-scale drug development that takes many years and comes at a steep price. Potential ways to cut consumer costs could include improving the first step, drug discovery. As computer models improve, predictions of leading compounds may become more accurate, resulting in less time and money spent on the first stage. SAR, mentioned earlier, will help identify potential mechanisms of action of the new compound; these predictions may aid in identifying side effects more quickly. Overall, this potential cost-savings from improved computational models will also reduce the total price of drug development and hopefully result in fewer failed compounds. Despite the amount of time and money it takes to advance a drug from the lab to your medicine cabinet, all stages of drug development are necessary to ensure safety and to advance human health.
 Jessica earned her BS in Biological Sciences from the University of Delaware. During her undergraduate, she conducted research on behavioral epigenetics and completed two pharmaceutical internships. Through these experiences, she became interested in epigenetics and disease, and she is now pursuing her PhD in pathology at the University of Michigan. Aside from learning, Jessica enjoys adding houseplants to her growing collection, exploring Michigan’s beautiful scenery, and driving her Mini Cooper.
Jessica earned her BS in Biological Sciences from the University of Delaware. During her undergraduate, she conducted research on behavioral epigenetics and completed two pharmaceutical internships. Through these experiences, she became interested in epigenetics and disease, and she is now pursuing her PhD in pathology at the University of Michigan. Aside from learning, Jessica enjoys adding houseplants to her growing collection, exploring Michigan’s beautiful scenery, and driving her Mini Cooper.

{kind=link}
Thank you for this post, it conatain a lot about drug discovery process and development. I hope you will share more.
LikeLike
very nice review on the drug discovery and development process
Great thanks
LikeLike